Optimizing Pharmaceutical Manufacturing: Addressing Muda, Mura, and Muri Through Modern Technology3/18/2023 
Md. Saddam Nawaz Head of Quality Assurance, ACI HealthCare Limited The pharmaceutical industry is one of the most complex and highly regulated industries in the world, with strict requirements for product quality and safety. The manufacturing of pharmaceutical products involves a range of processes, from research and development to clinical trials, production, and distribution. To ensure that products meet regulatory standards and are delivered to patients in a timely and efficient manner, pharmaceutical companies must address a range of challenges in their manufacturing processes. Three critical challenges that can impact manufacturing efficiency and effectiveness are Muda, Mura, and Muri. In the world of Lean Manufacturing, Muda, Mura, and Muri are three Japanese crucial concepts that help organizations identify and eliminate waste and inefficiencies in their processes. These concepts are equally applicable in the pharmaceutical industry, where quality and efficiency are of paramount importance. Muda Muda refers to any activity that does not add value to the final product. This could be a process step that is redundant, unnecessary, or does not contribute to the quality of the final product. Examples of Muda in pharmaceutical manufacturing include overproduction, waiting, unnecessary motion, excess inventory, over-processing, defects, and unused talent. Overproduction is a common Muda in pharmaceutical manufacturing. This occurs when production is not aligned with customer demand, resulting in excess inventory, waste of resources, and potential quality issues. To avoid overproduction, organizations can implement a pull production system where production is based on customer demand. In 2013, the US Food and Drug Administration (FDA) issued a warning letter to a pharmaceutical company for overproducing drugs without adequate controls in place. The FDA found that the company's manufacturing process resulted in unnecessary movement and transportation of materials, which contributed to delays in product release and increased the risk of contamination. The FDA also found that the company had not properly validated their manufacturing processes, which led to excess inventory and overprocessing. Waiting is another area where Muda can be found. This can include waiting for raw materials or equipment, waiting for approvals or in process check, or waiting for operators to be available. To address waiting times, organizations can optimize their production schedules, implement a visual management system, and prioritize tasks to ensure that production is continuous and uninterrupted. In 2019, the FDA issued a warning letter to a pharmaceutical company for excessive waiting times during the manufacturing process. The company had failed to implement a robust production schedule, resulting in operators waiting for equipment to be available, resulting in significant downtime. The FDA found that the waiting times had led to implement a more efficient production schedule. Excess inventory is also a common Muda in the pharmaceutical industry. This can lead to waste of resources, increased storage costs, and increased risk of quality issues. To reduce excess inventory, organizations can implement a just-in-time (JIT) system where inventory is only ordered and produced when needed. Mura Mura, on the other hand, refers to variation in the production process that results in inefficiencies, quality defects, and inconsistent output. Mura can arise from inconsistent workloads, variation in raw materials or equipment, or lack of standardized procedures. Examples of Mura in pharmaceutical manufacturing include fluctuating production volumes, inconsistent product quality, and variability in equipment performance. In 2018, the FDA issued a warning letter to a pharmaceutical company for variation in the quality of raw materials used in their drug manufacturing process. The company had failed to properly test the raw materials for quality and had used materials that did not meet the required specifications. The FDA found that the variation in raw materials had led to inconsistent product quality and ordered the company to improve their testing procedures. In 2017, the FDA issued a warning letter to a pharmaceutical company for failing to follow established procedures for drug manufacturing. The company had failed to properly train operators on the procedures, resulting in inconsistencies in product quality. The FDA found that the lack of standardized procedures had put patients at risk and ordered the company to implement proper training and procedures. In another warning letter, the FDA found that a pharmaceutical company's batch records were inconsistent, with missing or incomplete information. The FDA noted that the company did not have a standardized process for completing batch records, which led to variability in the quality of the products. Also, the FDA found that a pharmaceutical company's cleaning procedures were inconsistent, with different cleaning procedures being used for the same equipment. The FDA noted that the company did not have a standardized cleaning process, leading to variability in the cleanliness of the equipment and potential contamination of the products. To address Mura, organizations can implement adequate standard operating procedures (SOPs) and work instructions. A well written SOP can reduce variation in product quality, streamline production processes, and reduce the risk of quality defects. For example, SOP for cleaning equipment or conducting in-process checks can reduce variability and improve product quality. Another way to address Mura is to implement a Total Quality Management (TQM) system. This system aims to continuously improve the quality of products and services by involving all employees in the process of identifying and solving problems. By focusing on continuous improvement and standardization through internal audit, organizations can reduce risk, variability and improve the quality of their products. Muri Muri is the third concept in the Lean Manufacturing trifecta, and it refers to overburdening workers or equipment beyond their capacity. This can lead to employee burnout, safety concerns, and reduced efficiency. Examples of Muri in pharmaceutical manufacturing include expecting operators to run equipment for extended periods without adequate rest, pushing machines beyond their rated capacity, and demanding unrealistic production volumes. Additionally, if a manufacturing process is overly complex or requires excessive manual intervention, it may be difficult to manage and prone to error, leading to additional stress and inefficiency. This is also an example of Muri because the process is overly burdensome and difficult to manage. In 2016, the FDA issued a warning letter to a pharmaceutical company for violating in their manufacturing process. The FDA found that the company's equipment was being overburdened, causing unnecessary strain on the equipment, and leading to decreased reliability and quality. Specifically, the company was using equipment beyond its rated capacity, which led to frequent breakdowns and production delays. The FDA also found that the company's employees were being overworked, leading to fatigue and decreased productivity. The company had set unrealistic production targets and required their employees to work long hours without adequate rest, leading to burnout and safety concerns. The FDA concluded that the company's violations posed a risk to public health and required immediate corrective action. The FDA ordered the company to implement a Total Productive Maintenance (TPM) system to prevent equipment overburdening and to revise their production targets to reduce the burden on their employees. The company was also required to provide adequate training and resources to their employees to ensure their safety and well-being. The application of modern technology is a critical enabler for addressing Muda, Mura, and Muri in the pharmaceutical industry. By leveraging technology such as IoT, AI, and Big Data analytics, pharmaceutical companies can improve their manufacturing processes, reduce waste, and improve patient outcomes. By leveraging IoT and real-time data analytics, pharmaceutical companies can identify and eliminate waste in their manufacturing processes. For example, real-time data can be used to optimize machine utilization, reduce material waste, and improve quality control. AI and Big Data analytics can help in optimizing production planning and scheduling, thereby reducing variability in production processes. This can help in ensuring that the production process is smooth and stable, which in turn can lead to reduced waste and improved quality. The use of technology can help in preventing overburdening of equipment and employees. For instance, predictive maintenance using IoT sensors and machine learning algorithms can help identify potential equipment failures before they occur, thereby reducing equipment downtime and maintenance costs. Additionally, AI-powered scheduling and planning tools can help in optimizing workloads and preventing employee burnout. Here are some case studies of pharmaceutical companies that have implemented Industry 4.0 technologies to address Muda, Mura, and Muri: Pfizer's Digital Plant Initiative: Pfizer has implemented a Digital Plant Initiative that leverages Industry 4.0 technologies to optimize manufacturing processes, reduce downtime, and improve product quality and safety. The initiative involves the integration of digital technologies, such as real-time monitoring, predictive maintenance, and advanced analytics, with traditional manufacturing processes to increase efficiency and productivity. As a result, Pfizer has reported significant improvements in overall equipment effectiveness (OEE), reduction in downtime, and increase in production yield. Sanofi's Smart Packaging Initiative: Sanofi has implemented a Smart Packaging Initiative that utilizes Industry 4.0 technologies, such as IoT-enabled packaging and RFID tags, to track and monitor the supply chain of pharmaceutical products. The initiative aims to reduce waste, improve patient safety, and comply with regulatory requirements. As a result, Sanofi has reported a reduction in supply chain costs, improved visibility into the supply chain, and increased patient safety. Merck's Smart Manufacturing Initiative: Merck has implemented a Smart Manufacturing Initiative that leverages Industry 4.0 technologies, such as machine learning, artificial intelligence, and predictive maintenance, to optimize manufacturing processes and improve product quality and safety. The initiative involves the integration of digital technologies with traditional manufacturing processes to increase efficiency and productivity. As a result, Merck has reported significant improvements in manufacturing efficiency, reduction in downtime, and increase in production yield. Novo Nordisk's Cobots Initiative: Novo Nordisk has implemented a Cobots Initiative that utilizes Industry 4.0 technologies, such as collaborative robots (cobots), to automate manual tasks in the manufacturing process. The initiative aims to reduce the risk of human error, improve productivity, and enhance product quality and safety. As a result, Novo Nordisk has reported a reduction in errors, increased productivity, and improved worker safety. Overall, implementing Muda, Mura, and Muri concepts in the pharmaceutical industry can lead to significant improvements in quality, efficiency, and cost-effectiveness. By identifying and eliminating waste, reducing variability, and balancing production demand with capacity, organizations can improve their bottom line and provide high-quality products to their customers. By addressing these issues through the use of modern technology, pharmaceutical companies can improve their operations and remain competitive in the market. edit.
0 Comments
 The Japanese phrase "the real place" is known as "gemba." It refers to the actual location where value is created in business and manufacturing, such as a factory floor or a retail space. In order to find and remove waste, enhance processes, and add value for customers, it is important to concentrate attention on the front lines, where the work is actually done. Gemba is one of the most important ideas in lean management. Lean management is a way to improve processes that started in manufacturing but has since been used in a wide range of industries and organizations. The idea behind Gemba is to go to where the work is being done, watch what's going on, and look for ways to make things better. By focusing on the front line, where the value is created, organizations can learn more about their processes, find areas of waste and inefficiency, and make changes that improve quality, boost productivity, and make the customer experience better. The Gemba walk is a key part of putting the Gemba concept into action. It means going to the front line and observing and talking to employees, as well as collecting data and information to find places where things could be better. This process can be done over and over again to track progress and make sure that things keep getting better. The Gemba approach can not only improve processes and create value for customers, but it can also create a culture of engagement and empowerment by getting employees involved in the process of improvement. By having employees take part in the Gemba walk and giving them a say in the process improvement effort, organizations can build a strong sense of ownership and commitment to quality and continuous improvement.The Gemba walk is a structured process that typically involves the following steps:
It's important to note that the Gemba walk is an ongoing process, not a one-time event. Organizations can maintain a focus on continuous improvement and drive meaningful results by visiting the front lines on a regular basis, observing and collecting data, analyzing and improving processes. Post-Gemba activities follow the Gemba walk, which is a structured process used to improve processes and create value for customers. Most of the time, the post-Gemba process involves analyzing the data and information gathered during the walk, figuring out what needs to be improved, and coming up with and putting into action ideas and plans for improvement. The post-Gemba process is an important part of the continuous improvement cycle because it helps to make sure that the changes made during the Gemba walk are having the desired effects and that continuous improvement is being kept up. The steps involved in the post-Gemba process typically include:
By continuously visiting the front line and following up on improvement efforts, organizations can maintain a focus on continuous improvement and drive meaningful results over the long-term. *This article was originally posted on www.linkedin.com
Md. Saddam Nawaz Head of Quality Assurance, ACI HealthCare Limited  Image Source: freepik.com Vaccines are time- and temperature-sensitive pharmaceutical products (TTSPPs). Some vaccines are susceptible to freezing, whereas others are sensitive to heat and light. Vaccines must be shielded from temperature extremes in order to maintain quality. Most widely used cold chain vaccinations, for example, are kept between +2 and +8 °C, or around the temperature of a standard refrigerator. Some vaccines, such as Modena's COVID-19 candidate and the varicella vaccine for chickenpox, must be kept frozen at -20 degrees Celsius. A few, must be stored at extremely low temperatures and require what is known as the ultra-cold chain consistent storage at -75 °C. Thermostability was always going to be an issue for early distribution of TTSPPs, where the key challenges are Electricity (Ultra-cold freezers in hot places require even more power), cold-chain storage challenges (candidates may requires multiple doses), and distribution/transportation time (journey from the factory to the point of care takes months). Therefore, aside from the technical issues, the manufacturer, importer, distributor, and medical institutions will need to collaborate to reduce the chance of failure and ensure patient safety. The goal of this article is to identify common risks associated with the storage and transportation of time- and temperature-sensitive pharmaceutical products (TTSPPs), such as temperature-sensitive small molecules, vaccines, biologics, biotechnological products, radiopharmaceuticals, and combination products, as well as to suggest mitigation strategies. The purpose of this article is not to prescribe specific procedures or to address present regulatory frameworks; rather, it is to focus on quality process risks and mitigation strategies to maintain product and supply chain integrity. A strategy for assessing, controlling, communicating, and reviewing risks identified at all levels of the supply chain should be in place. The risk assessment should be founded on scientific knowledge and familiarity with the process, and it should finally be tied to the patient's protection. Product and process information is the beginning point for recognizing hazards associated with drug product storage and transportation. Process mapping is a useful tool for businesses to acquire a better knowledge of a specific process and/or operation (e.g., transport lane selection or warehousing and transport loading/unloading patterns). Table 1: Potential major risk areas and stakeholders in the supply chain  The risk assessment table above is applicable to companies and persons involved in the storage and transportation of TTSPPs , including but not limited to:
QUALITY MANAGEMENT SYSTEM (QMS) Quality Management System (QMS) describes a company's procedures with an emphasis on achieving the needs and expectations of its customers. The QMS should include the organizational structure, procedures, processes, and resources, as well as the actions required to assure confidence that the delivered TTSPP retains its quality and integrity and continues inside the legal supply chain during storage and/or transportation. Senior management is ultimately responsible for establishing, effectively funding, implementing, and maintaining a successful quality system. Throughout the organization, the effectiveness, roles, duties, and authorities should be defined, communicated, and implemented. The QMS should ensure the following:
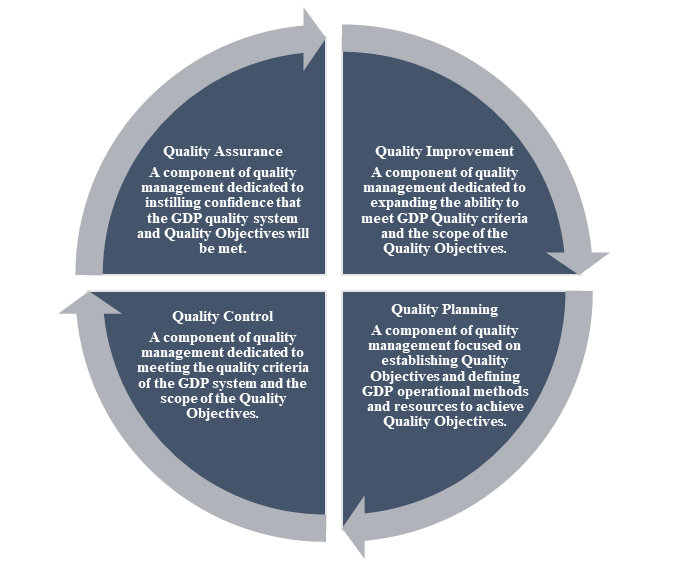 Figure 1: QMS in Good Distribution Practice The QMS is a management tool that helps the company meet regulatory, customer, and business needs. As a result, management should review the QMS to ensure it is still relevant. Management should have a defined process for periodically reviewing the quality system. Monitoring the achievement of quality system objectives. PERSONNEL Most supply chains are complex. Therefore, Individuals who verify, record, pick, transport, and deliver TTSPPs must be trained and competent to respond effectively to information that may affect product quality. Companies must have enough staff or responsible persons familiar with the legislation to implement and oversee the requirements. Personnel appointments should be scrutinized. Some significant areas of knowledge and experience include:
All distribution personnel should receive GDP training. They should be knowledgeable and experienced before starting their tasks. Initiate and maintain role-specific training based on documented procedures and a documented training plan. Regular GDP training should keep the accountable person current. Furthermore, training should cover product identification and how to avoid falsified medicines entering the supply chain. Appropriate personnel hygiene procedures, relevant to the activities being carried out, should be established, and followed. PREMISES The facilities should be designed or modified to ensure that the required storage conditions are met. They should be sufficiently secure, structurally sound, and large enough to allow safe storage and handling of the drug products.
MONITORING DEVICE The shelf life of a drug is determined by its chemical and physical properties, as well as the storage and transportation temperatures. Monitoring those conditions is critical in TTSPPs transportation and storage. Temperature-controlled rooms, cold rooms, refrigerators, and freezers used to store TTSPPs should have air temperature monitoring systems and devices. Follow the bare minimums:
TRANSPORT The supply chain for time- and temperature-sensitive pharmaceutical products (TTSPPs) should be secure enough to prevent product theft and reduce the risk of falsified products entering the supply chain. The chosen transportation route should ensure that the products are always kept within the required temperature ranges. Documented evidence of the supply chain route that the products were distributed through and the conditions to which they were exposed during transportation should be kept and made available. Temperature monitoring in transports via data loggers included in the shipment or prior qualification of the transportation route should be used to demonstrate this (considering seasonal variations). All product distribution should be based on a risk assessment and documented appropriately, taking into account the various factors listed in Table 2 that may affect the products. Table 2: Example of a risk-based approach to planning transportation 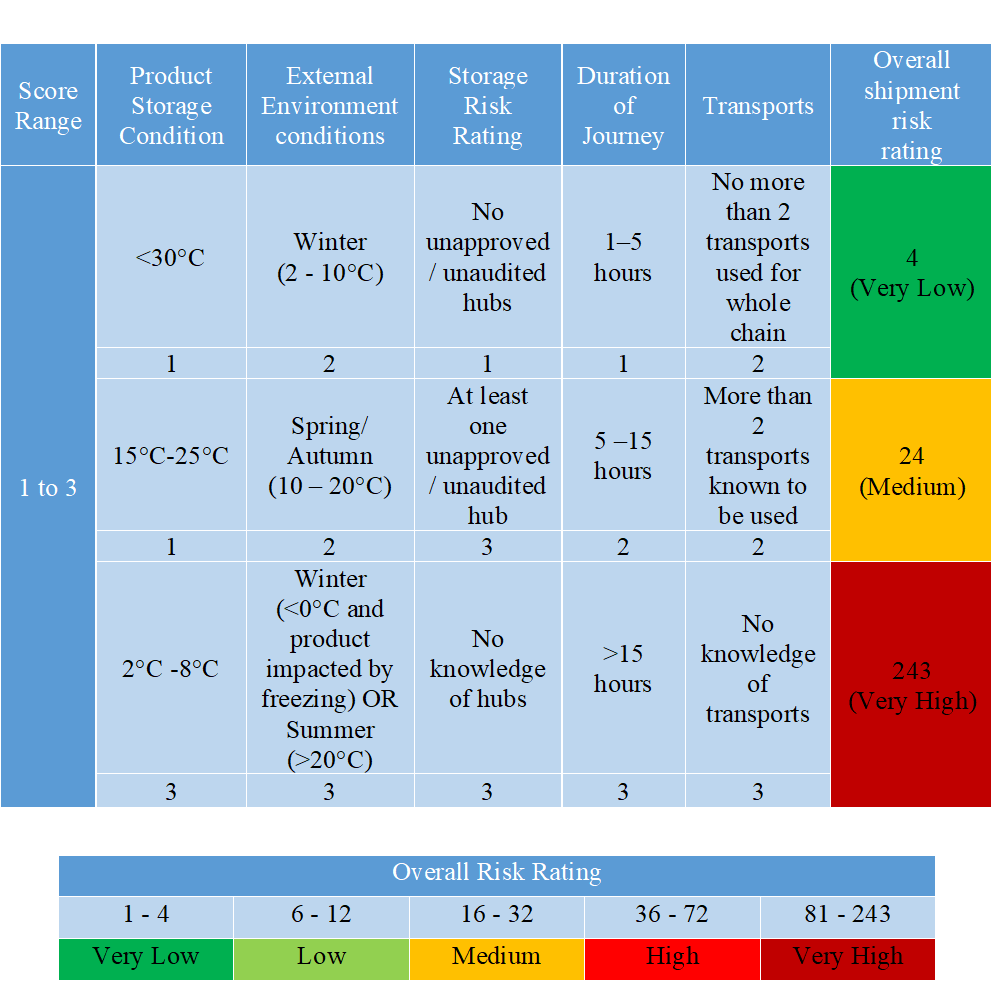 The storage and transportation processes for TTSPPs may involve complex movements with differences in documentation, handling requirements, and communication between the various entities throughout the supply chain. Drugs must be stored and transported according to predetermined conditions (e.g., temperature) as supported by stability data. Temperature excursions outside of their respective labeled storage conditions, for brief periods, may be acceptable provided that stability data and scientific/technical justification exist, demonstrating that product safety, quality, and efficacy is not affected. If a deviation such as temperature excursion or product damage has occurred during transportation, this should be reported to the distributor and recipient of the affected drug products. A procedure should also be in place for investigating and handling temperature excursions.
COMPLAINTS
It is mandatory to meticulously document and handle all complaints. Complaints should be documented in full. Complaints about TTSPP product quality should be distinguished from complaints about distribution. In the event of a product defect, the manufacturer and/or regulatory body should be notified immediately. Any product distribution complaint should be thoroughly investigated. Appoint someone to handle complaints and provide adequate support. After the investigation and evaluation of the complaint, appropriate follow-up actions (including CAPA) should be taken, including notification to national competent authorities. Wholesale distributors must immediately notify the competent authority and the holder of the marketing authorization of any suspected falsified drug products. A procedure should be in place. It should be documented and investigated. Falsified drug products should be physically separated from other drug products and stored in a separate area. All relevant product activities should be documented and kept on file. The effectiveness of product recall plans should be evaluated regularly (at least annually). Records should be easily accessible to authorities. Distributor and directly supplied customer information (addresses, phone and/or fax numbers, batch numbers at least for drug products bearing safety features as required by law, and quantities delivered) should be readily accessible to the person(s) responsible for the recall. A final report should track the recall's progress. REFERENCES
 The firm failed to establish an adequate system for monitoring environmental conditions in aseptic processing areas (USFDA 21 CFR 211.42(c)(10)(iv))
The firm produces (b)(4) injection (b)(4) mg/vial for the US market. The USFDA inspection revealed deficiencies in the aseptic processing facility's ongoing monitoring and control. Failure in pressure differentials monitoring The firm discovered that it lacked an efficient method for ensuring appropriate differential pressure management in its aseptic processing plant. During a (b)(4), they manually recorded differential pressures between cleanrooms (4). They also permitted pressure reversals and excursions up to (b)(4). Notably, for deviations/incidents that exceed (b)(4), a deviation/incident “may” be recommended. They also lacked a sufficient, integrated system for monitoring room differential pressure on a regular basis during a (b)(4), examining continuing HVAC control, starting deviations, and taking necessary action when pressure excursions occurred. They lack evidence that ongoing control was maintained for the HVAC systems used in their aseptic processing operations due to insufficient pressure differential systems and insufficient standards for initiating investigations of pressure control issues. To maintain appropriate air quality throughout all of their cleanrooms, a suitable facility monitoring system is essential. All alarms and deviations from established limits should be thoroughly investigated in order to detect atypical changes that could jeopardize the facility's environment as soon as possible. Prompt detection of an emerging low-pressure problem is critical for preventing lower quality air from entering a higher criticality room. The firm’s response stated they are implementing a new system that focuses on monitoring online differential pressures for the (b)(4) (filling line). However, they did not commit to a comprehensive review of their facility monitoring and HVAC systems to improve environmental control in all cleanrooms of the aseptic processing facility. Failure in non-viable particulate monitoring The firm lacked an adequate system for handling non-viable particulates (NVP) above their action level of (b)(4) NVP ≥ 0.5 μm/ftᶾ during aseptic processing operations. This limit was exceeded for seven out of (b)(4) batches of (b)(4) injection (b)(4)mg/vial manufactured since May 2019. The firm lacked investigations in response to the high particulate levels observed in the ISO 5 aseptic processing operation. Per their NVP monitoring procedure, an investigation was not required unless an excursion lasted longer than (b)(4). Excessive particulates in the ISO 5 environment can lead to nonviable or biological contamination of sterile drug products. In their response, they stated that they are in the process of evaluating the feasibility of implementing an integrated non-viable particulate counter within the (b)(4) which will enable the filling machine to stop when an excursion occurs. Although they have committed to improvements in their non-viable particulate monitoring program, they lacked sufficient information on the parameters that trigger investigations. In response to the letter from the firm, the agency seeks the following: A thorough, independent assessment, and CAPA for their pressure differential system. Include a comprehensive evaluation of monitoring, recording, alarm documentation, deviation investigation, data retention and overall system control in their assessment. CAPA should includes but is not limited to:
A Comprehensive risk assessment of all contamination hazards with respect to the firm’s aseptic processes, equipment, and facilities. The firm must provide an independent assessment that includes, but is not limited to:
A detailed remediation plan with timelines to address the findings of the contamination hazards risk assessment. The firm shall describe specific tangible improvements to be made to aseptic processing operation design and control. Finally, the firm should see FDA’s guidance document, Sterile Drug Products Produces by Aseptic Processing – Current Good Manufacturing Practice, to help you meet the CGMP requirements when manufacturing sterile drugs using aseptic processing, at https://www.fda.gov/media/71026/download. Note: (b)(4) = confidential corporate information
Md. Saddam Nawaz Head of Quality Assuracne, ACI HealthCare Limited  An FDA inspector has been at your facility for several weeks or longer, and that the inspector has issued a form 483 and this is a significant compliance situation that will likely result in further FDA action. Which course of action should you take now? The FDA currently has a policy identical to that of a Warning Letter, requiring a response within 15 business days of receipt. Policy published on August 11th, 2009, in the Federal Register Vol. 74 No. 153 indicates that the FDA intends to conduct a comprehensive examination of the response if it receives it within 15 business days after the 483 was issued. To resolve legislative or regulatory breaches, the Warning Letter was introduced. Warning Letters are only issued in the event of serious regulatory violations, according to agency regulation. Significant violations are those that may result in enforcement action if they are not promptly and adequately corrected. When it comes to enforcing the Federal Food, Drug, and Cosmetic Act, the agency's principal instrument is a Warning Letter. As a result, it is plainly desirable for a company to submit the 483 responses on time for a variety of reasons, including: - It gives the investigated organization a chance to express themselves. - Consequently, the FDA must take a little more time to think about their stance. - An eventual Warning Letter might be postponed. - It gives the investigated organization a chance to explain their CAPA (Corrective and Preventive Action) strategy. - However, the FDA will offer more specific recommendations on what extra remedial steps are needed if a Warning Letter is issued. - To the agency, it indicates that the investigated organization is serious about the inspection. When responding to a 483 or a Warning Letter, there are several factors to consider. The basic rule is to be direct, honest, and concise, and to concentrate solely on the FDA's findings. Remediation is generally handled by a dedicated team of people, often managed by someone from the firm's regulatory affairs or quality assurance departments or compliance. To demonstrate the company's commitment to a successful response, executive management should be involved from the start. There are several layers of skilled FDA employees that will evaluate the answer. They are skilled at spotting superficial answers that boast self-serving compliments for aesthetic purposes. Such a reaction will do more harm than good. You are not required to confess wrongdoing or to embarrass yourself or your company, but you must step up to the plate, offer good recorded CAPA proof, and be fully prepared to see it through. Overall, FDA is seeking solid proof that the same or comparable defects will not recurrence. There are many of the same things that go into a Warning Letter answer that also apply to the 483. CAPAs should include references to and attachments of supporting documents, such as approved Standard Operating Procedures, documented training of personnel, and any additional steps that have previously been taken or will be implemented in the future, with completion dates if necessary. FDA inspections of previous operations that were utilized or completed some years ago are not unusual. Many remedial steps may have already been implemented in this situation, easing FDA worries for future operations. However, you must have strong proof and paperwork to back up your claim, which must be linked to your answer. As part of its inspection, the FDA may have also looked at a new facility and found nothing abnormal. You could note in your response that this is the best proof of rectification available, as it has been confirmed by FDA. Formatting the response should focus on the 483 observations, and it is typically recommended to reiterate each observation word-for-word before providing your response. There are several exceptions to this technique when there are numerous observations pertaining to the same basic shortcoming or when there are so many specifics that the reader may become confused if the observation is not simply stated. The difficulty of responding to some 483s depends on the inspector's level of experience, and this is especially true in GMP. If you want to stay on track and on target, it's a good idea to summarize or paraphrase the FDA's observation and then respond to it. On the other hand, you do not want to go overboard and provide so much information that it takes time and is difficult to evaluate. Note that everything you provide to the FDA is also "inspected" in detail. If you reply to a Warning Letter that calls for the agency to be closing, all quantities of extra materials will lengthen the time it takes FDA to complete its examination and can thus be counterproductive. The 483 response is slightly different since the inspected company will benefit from the additional FDA review time. However, the answer is focused on the observations followed by recorded corrective measures. When assessing the 483 and choosing the best method to take, keep in mind that common sense should be applied in each instance, and professional judgment is important. While no two circumstances are the same, there are certain general guidelines that may be applied to all or any of them:
You're not compelled to respond to inspectors' Form 483 observations. It is, however, not suggested to do so. If you don't respond to the FDA's concerns in the 483 observation, you're much more likely to receive a warning letter. If you receive a warning letter, you are required by law to make any necessary modifications to address the FDA's concerns. A 483 observation is significantly less serious than a warning letter. Any breaches must be addressed before you can meet compliance and commercialize your product. Finally, you want to ensure that your company can complete what it promised in its FDA response, considering the practicalities of each item. In any inspection, the FDA will focus on violations of their regulations; the general purpose of the response is to give compelling evidence that the same or identical deviations will not occur again in the future. *This article was originally posted on www.linkedin.com
 Regulatory authorities are engaged in numerous activities to protect and promote public health during the COVID-19 pandemic, ranging from the acceleration of development for treatments for the novel coronavirus, maintaining and securing drug supply chains, providing guidance to manufacturers, advising developers on how to handle clinical trial issues and keeping the public informed. This article examines the guidances published by the UK Medicines and Healthcare products Regulatory Agency (MHRA) on Inspections and good practice that can be applied to help pharmaceutical companies cope with the Good laboratory and Distribution Practice consequences of the COVID 19 pandemic, while ensuring a high level of quality, safety and efficacy for medicinal drugs made available to patients. Guidance 1: Approval of GxP documents when working from home during COVID-19 outbreak Due to the COVID-19 pandemic, remote or home-based working has increased. This guidance facilitates organisations to consider alternative methods whilst maintaining basic control of GxP documents. GxP documents can generally be spilt into two categories:
Conversely, documents that have traditionally been printed on paper, physically handed to each reviewer and/or approver in an office and authorised using wet ink signatures can be shared with remote workers, but they have no formal system describing how the review and approval can be recorded. Such documents include, but are not limited to, validation protocols and reports, risk assessments, technical reports, quality management system documents that are paper-based such as standard operating procedures (SOPs), investigations and change requests etc. Therefore, the MHRA recommends identifying the aspects to consider with remote approval, including:
The guidance urges that the solution will vary from firm to firm, depending on the type of document and the tools available to the person performing the approval – eg, printer, scanner/smartphone, secure email, third party software or existing systems that have tools to capture electronic signatures. No matter what system or process is used, the following principles should be applied to maintain control:
Guidance 2: Guidance for manufacturers and Good Practice (GxP) laboratories on exceptional flexibilities for maintenance and calibration during the coronavirus outbreak The agency is allowing alternative courses of action for manufacturing or laboratory equipment during the outbreak. The following options are available to manufacturers and GxP laboratories to conduct maintenance and calibration during the COVID-19 pandemic. If an engineer is available to attend site An organisation’s health and safety procedures or manual should be updated so the engineer can ensure appropriate social distancing measures are upheld and be supervised adequately during their time on site. Calibration and maintenance protocols or similar documents may be reviewed prior to the engineer visiting the site as per routine requirements. Approval may be given electronically and the site visit documentation may also be signed electronically. If an engineer cannot attend site but is available by telephone or video call A qualified employee may perform the calibration or maintenance task under remote supervision from the engineer, providing the site has all the required materials, parts and tools to perform the task. If an engineer is not available to attend site and remote supervision is not possible A pharmaceutical quality system record (eg, change control or deviation) should be raised and the delay to the calibration or maintenance task should be risk assessed, considering;
Off-site calibration/maintenance Navigable equipment can be shipped off-site for calibration or maintenance. If this is not standard practice then the firm should detail the risks of equipment leaving the site for calibration or maintenance and examine whether it is fit for the intended use after return to the site. Substitution of laboratory equipment Any analytical methods that have been validated/verified on a specific make/model of equipment can be transferred to an equivalent instrument after assessment of the impact on the validated/verified status of the method. If required, method validation/verification may need to be performed again to ensure the validity of the results generated. Guidance 3: Guidance for Good Laboratory Practice (GLP) facilities in relation to COVID-19 The UK Good Laboratory Practice Monitoring Authority (UK GLPMA) recognises there are potential challenges that COVID-19 will present to members of the GLP monitoring programme. Office-based GLP inspections Office-based inspection can be conducted and once the inspection is closed a statement of compliance will be issued to provide that facility with a record of continued membership of the monitoring programme. However, an onsite facility inspection will then be scheduled once the regular inspection programme resumes. Amendments and deviations Any incident or deviation encountered due to COVID-19 that could potentially impact the GLP status of a study should be maintained, fully assessed and documented via existing amendment and deviation programme by the firm. Quality Assurance (QA) The guidance recommends that QA activities are prioritised using a risk-based approach. A risk assessment can be used to identify where to focus resources and adapt audit programmes. The impact of deviation from planned QA audits or delays due to a lack of resources or travel restrictions should be assessed and documented by the study or quality director. Remote technology such as video calls can be an alternative to physical presence and would be acceptable. The use of remote observation methods should be fully risk-assessed to ensure they provide a similar level of oversight as a physical audit. Guidance 4: Exceptional good distribution practice (GDP) flexibilities for medicines during the COVID-19 outbreak The MHRA’s temporary regulatory flexibilities address the current exceptional circumstances. However, they are being regularly reviewed and may be updated at any time. Supply chain Periodic supplier and customer requalification may be postponed. Interim reliance may be placed upon regular review of notifications of suspended wholesale dealer authorisations (WDA(H)) and any General Pharmaceutical Council registration updates. Medicines may be returned to saleable stock if returned from the wholesale distribution chain within 10 days. Transportation Non-temperature-controlled transport may be used when the ambient temperature is less than 20°C. A risk assessment should be in place for products transported under less than 8°C, with justified mitigating measures and with ‘Do not refrigerate’ identification. Another risk assessment ought to be conducted for products that can be held for up to 96 hours at a transit hub without a WDA(H) to assist in transportation. Alternative arrangements to show proof of delivery will be permitted. Responsible Persons (RP) RPs may fill in for RPs from another company, providing the companies are grouped without variation and have an RP registration number issued by the MHRA. Facilities and equipment Storage and distribution equipment may be used as soon as possible, supported by a risk assessment and additional mitigating measures with limited qualification and validation. Remaining qualification and validation work should be completed retrospectively, with any delays minimised.
The MHRA recognises that the care of patients is every firm’s priority. To achieve this goal and ensure only the highest quality products are manufactured, the MHRA requires that firms make relevant decisions which have been carefully evaluated for risks. *This article was originally posted on www.europeanpharmaceuticalreview.com
 The rise in Data Integrity warning letters are forcing companies to make a beeline for obtaining an understanding of data integrity. A close examination of management objectives where data integrity issues have unraveled indicates that they have been driven by the self-interest of profit. They hesitate to switch out older equipment for newer ones with technical controls to enforce data integrity. They also hesitate to provide the required level of personnel resources for regular audit trail reviews, investigation of data integrity issues etc. While regulatory agencies are actively hiring computer savvy personnel familiar with the intricacies of electronic data, business expediency dictates pharmaceutical industry management to shadow those efforts by ensuring that adequate budgets are allocated to hire personnel with the right blend of IT and compliance expertise. The purpose of this paper is to suggest a control framework to ensure data integrity in an organization. Contaminated data MHRA’s July 2016 draft version for GxP Data Integration Definitions and Guidance for Industry defines data integrity as “the extent to which all data are complete, consistent and accurate throughout the data lifecycle.” We may consider data integrity as synonymous with product purity wherein the product is either contaminated or not contaminated. So too with data integrity where the metric is binary in nature. Data is either contaminated or not contaminated. There is no in between to signify a “degree of breach or contamination”. So what is data integrity? Data Integrity may be appropriately defined as “the state of completeness, consistency, timeliness, accuracy and validity that makes data appropriate for a stated use”. It is a data characteristic that lends it the assurance of trustworthiness. It is defined by the oft-mentioned ALCOA+ attributes. NIST SP 800-33 defines data integrity as the state when data has not been altered in an unauthorized manner. It covers data in storage, during processing and while in transit. Data integrity’s guiding principles include:
Assuring enterprise wide data integrity When it comes to assuring data integrity, the situation is more complex because words mean different things to different people. To the IT Security group it is the assurance that information can be accessed and modified only by those authorized to do so. To the Database Administrator it is about data entered into the database are accurate, valid and consistent. To the Data Owner it is a measure of quality, with existence of appropriate business rules and defined relationships between different business entities and to the Regulator, data integrity is the quality of correctness, completeness, wholeness, soundness and compliance with the intention of the creators of the data. This difference in meaning creates a fertile ground for miscommunication and misunderstandings, with the risk that the activity will not be done well enough because of unclear accountabilities. Notwithstanding the impossibility of eliminating all vulnerabilities to data integrity in the organization, controls should be established to reduce the propensity for data integrity errors and vulnerabilities. Such controls should integrate and coordinate the capabilities of people, operations, and technology through a data integrity assurance infrastructure. It hinges upon a multi-faceted approach consisting of the following triad components:
Management controls Management controls address the people and business factors of data integrity. They describe the means by which individuals and groups within an organization are directed to perform certain actions while avoiding other actions to ensure the integrity of data. These controls are enumerated in the company’s Ethics Policy, Code of Conduct directive, Data Management and Governance program etc. They serve as the enabler of a collaborative approach for governance of data that affect product quality and patient safety. The four key areas that management controls should address are:
Control environment is the establishment and maintenance of a working environment also called the company’s culture. It is the declaration of the company’s ethics and code of conduct for all employees. Ethics is management’s declaration of their moral values and philosophy. Code of Conduct is a set of rules according to which people in the company are supposed to behave and the consequences they would have to face for failure to do so. Control activities are captured in a Data Management and Governance program. It includes items such as data ownership, data stewardship, roles and responsibilities of different groups, risk assessment and alignment, controls performance metrics etc. Information and communication is management’s commitment to encourage all to communicate data integrity failures and mistakes. This may be accomplished through a regularly scheduled reporting mechanism wherein stakeholders receive reports on data integrity controls’ Key Performance Indicators (KPIs). These reports enable stakeholders to direct their business to achieve the desired data integrity goals. The KPI details, frequency of issuing reports, typical report contents etc. are some of the items that Data Governance program should identify. Monitoring is one of the most critical but often misunderstood process. It involves regular reviews of performance of data integrity. It helps to identify and remediate deficiencies in the controls. It also determines if and when data integrity directives need modification in order to meet changing business needs. Procedural controls Procedural and administrative controls are guidelines that require or advise people to act in certain ways with the goal of preserving data integrity. They are embedded in the company’s core business activities. They consist of a suite of approved documents that provide company personnel specific directives for activities that preserve and protect the integrity of data. These controls fulfill the ALCOA+ dimension of data integrity. Technical controls Technical controls are controls or countermeasures that use technology-based contrivances in order to protect information systems from harm. These can include mechanisms such as passwords, access controls for operating systems or application software programs, network protocols, firewalls and intrusion detection systems, encryption technology, network traffic flow regulators, and so forth. When used together, the adoption of these different types of controls allows for the establishment of a layered defense, and provides the best chance possible of preventing data from integrity breaches. Whereas procedural control is primarily applied to practices and procedures during data’s lifecycle, technical control is designed into products to preserve data integrity during the three data states, which are as follows:
Some consider 21 CFR Part 11 and Annex 11 regulations as the “data integrity” regulation. This is partially true. These regulations primarily address the technical controls that address data security. They should be designed into products and include features like role-based access control, audit trail capture and storage, capability to accept global reset of clock time etc. Design consistency, an ALCOA+ attribute, is also a manifestation of technical control. An example of such a realization of consistency is through an enterprise IT architecture design centered on a SOA (service-oriented architecture). Such a design provides a common standard for data interchange and along with a common information model provide the boost to data consistency during “data in motion” state. Besides system architecture, design controls are also realized via Master Data Management (MDM), Common Data Model (CDM). Data integrity benefits accruing from a consistent design philosophy enhances data integrity in the following ways:
Management’s pivotal role for triad’s success Data integrity efforts’ success in an organization is largely dependent on it’s culture. Since executive management primarily influences company culture, it is their ultimate responsibility to ensure data integrity. They should not delude themselves into a false sense of complacency and pretend they do not have any data integrity problem. Instead, they should prioritize their company’s efforts towards establishing a data integrity infrastructure and provide the leadership by allocating necessary funds and resources to developing the infrastructure. In addition to the leadership role that Executive Management provides, they also serve as the driving force for developing and implementing management controls while delegating the leadership role to middle level management to develop the procedural and technical controls. When establishing management controls, they should seek the expertise of outside consultants. These consultants provide the data integrity expertise along with valuable external perspectives on the company’s dynamics that can be difficult to see from the inside. The consultants also help in negotiating differences of opinions among team members by providing their valuable opinions based on their experiences with other companies. Executive management should also recognize the contributions of all their employees. As a result, they should empower them with decision-making and encourage them to be critical and divergent thinkers. Towards that end, they need to provide the necessary funding and hire the right people to develop the procedural and technical controls while they and their consultants develop the management controls. Conclusions A pharmaceutical company’s supply chain consists of business processes that produce and use regulatory data. Consequently, the triad controls along with the company’s Quality Management System (QMS) are applicable to all these processes. Hence, business expediency dictates that the triad controls are integrated with the QMS. Accordingly, the same managers who are responsible for day-to-day operations and product quality decisions should also be responsible for ensuring the integrity of data they use or create. The triad controls should be designed to complement one another with no overlaps. Since management controls consists of a Data Governance program, which is the company’s overarching program to ensure data integrity, the program elements must be traceable to the procedural and technical control elements. This triad inter-dependency is the key to ensuring an effective data integrity assurance infrastructure. The developers of triad controls cannot effectively judge the data integrity safeguards that the controls represent. Experienced data integrity consultants routinely find issues with the controls that are unknown to management or has eluded their attention. Nor were these detected during management oversights and internal audits. A fresh set of eyes of experienced consultants could significantly enhance the assurance of trustworthiness of data that triad controls are designed to achieve. *This article was originally posted on www.linkedin.com
 It is essential that management develop, document and implement procedure(s) for managing inspections by Regulatory Authorities in order to protect the legal rights of the business (and the Regulatory Authorities to perform repeated inspections) and at the same time, to maintain a professional relationship with the regulatory authority conducting the inspection. Senior Management at a site or function must be present during key parts of an inspection. Inspection notification, ongoing highlights of the inspection, and the final results of an inspection must be communicated to relevant Senior Management in a timely manner. There must be a local SOP describing the actions and responsibilities associated with an inspection. The SOP must address the local legal requirements for taking photographs; the use of tape recorders or other electronic equipment; the listening to, reading and signing of affidavits by company personnel; the review of internal audit reports and allowing access to computer databases. An accurate and detailed record is to be maintained of events, significant comments or recommendations from the inspector(s), documents and / or reviewed/copied for the inspector, product samples, and any other information deemed important to the inspection. At the conclusion of an inspection, local management must assure that any inspection observations are clearly understood, that any factual errors are brought to the attention of the inspector(s) and that there is a clear understanding of what will be addressed by the site in written communication to the Authority. Following a detail procedure that can be followed by any GMP site for preparation of inspection by regulatory authority. The purpose of the procedure is to define the procedure for the conduct of GMP sites employees involved in regulatory authority inspections. This procedure shall be used to guide GMP site personnel in contact or involved with the assistance of any external third party regulatory authority inspection. It is the responsibility of the Management Representative to implement and maintain this procedure. Preliminary meeting: The Inspector(s) and the responsible person are required to attend the preliminary meeting. Representatives from the following departments may be requested to attend the preliminary meeting if required:
The following information shall be provided at the preliminary meeting if requested: Company organization structure
The following shall be agreed during the meeting:
Inspection: During the inspection, the responsible person shall:
Daily Review Meetings:
Final Inspection Review Meeting: On completion of the inspection, a review meeting shall be held involving the inspector(s), the responsible and other appropriate personnel at which:
The responsible person must decide whether to:
Closing an Inspection:
 Reason # 1 Ductwork is not reinforced for the proper SMACNA (Sheet Metal and Air Conditioning Contractors' National Association) pressure classifications.  Reason # 2 Failure to adequately seal ducts.  Reason # 3 Failure to understand the need to seal return and exhaust ducts. Reason # 4 Failure to adequately pressure test ductwork to prove that duct sealing is effective.  Reason # 5 Failure to understand SMACNA Duct Construction Standards in the fabrication and installation process. Reason # 6 Failure to properly install turning vanes. Reason # 7 Duct velocities approaching 2500 fpm may create noise and will cause increased pressure drops in fittings and ductwork. Reason # 8 The detrimental impact on fan capacity caused by fan system effect is a problem not fully understood by the design industry. Reason # 9 Drawings with missing dimensional sizes are usually interpreted improperly. Undersized ductwork is usually installed. Reason # 10 Most specifications and details call for four duct diameters of straight duct before the inlet to a VAV (Variable Air Volume) box, but the actual duct drawings do not have sufficient room to allow for this installation. References *This article was originally posted on www.linkedin.com
 The term or function “Audit Trail” is very often totally misunderstood or misinterpreted. It gets even worse if the definition or understanding of the “Audit Trail” is not clear when the question about the “Audit Trail Review” arises. This might then end in bizarre and meaningless discussions without any solution. As mentioned in my most popular article "FDA External Audit Checklist" the CFR Part 11: “Audit trails” are frequently cited in FDA enforcement actions. It is important to remember that audit trails in electronic records are the equivalent of the “line-out, initial and date, explain” process used to identify and correct mistakes made in paper records. In the absence of appropriately configured and enabled audit trails it is impossible for a reviewer or auditor to ensure the data is valid and has not been altered or deleted. Warning letters have been issued for permitting conditions to exist where data may be changed or deleted. This publication intends to provide best practice insights into the misty world of Audit Trails. ✔ Fact #1: There is no clear definition of the Audit Trail function in general. Other industries are also using Audit Trails or IT developers might have another understanding of this function. There the Audit Trail is just seen as a simple log mechanism, tracking who has changed what and when. The GMP Audit Trail also requires the reason of change (not a description, not a comment, the real reason WHY it was changed). Logically the reason for change must be entered by the operator manually for each executed data change. ✔ Fact #2: If we talk about the Audit Trail we see it as a general / umbrella term. One of the first questions must be if we call / define an Audit Trail for the initial entry by the user or the first change of an initial entry. Again logically the first / initial entry by the user must be confirmed (e.g. by pressing the OK button, Return key etc.). ► From a GMP perspective the initial entry must not be audit trailed, because it must be recorded and documented anyway (what we always did). And it would make no sense to enter the reason for change, because it is not a change of the data (instruction type or record/report type). If it is not “audit trailed” (respectively not mandatory) an “Audit Trail Review” of the records does not really make sense. Or we mistrust the operator? Or we have no confidence on the process and data created – this might happen, if the process design is weak (retesting possible and not traceable) and not restricted by proper user access and rights / groups management (segregation of duties). But then an “Audit Trail” might not be the appropriate solution for it and re-design of the process would be required. ✔ Fact #3: A lot of vendors defined any kind of existing log or trail functions as Audit Trail, without a pre-defined basis of specification or interpretation All “Audit Trails” are identical? ► We prefer to call this type of “Audit Trail”, if you still like the general term, as the activity log or trail [Activity Trail]. For example this Activity Trail records all initial entries of the user (operator/analyst) which are part of the ordinary / normal work execution for each work step in a sequence of actions, comparable to a normal checklist on paper (protocol form, for each step one initial given). This Activity Trail might be very useful to replace each confirmation given by an initial on paper for each work step. This can also reduce the need giving tons of electronic signature for each step executed, if for example a single-sign on mechanism is used (based on login by user and automated log-off function). Admin = Admin? ► To make life easier, a second type of “Audit Trail” should be defined as Security Trail [Security Trail]. For that it must be clear, that the also very generally used term of the administrator role must be more precisely defined. You might find statements, that an administrator of a system can add/change users, assign users to user groups, etc. but also can change system configuration settings, even able to change methods or receipts (programs, calculations parameters) or even change network / server settings. If so, this is too much power concentrated one on single role. It makes sense to separate these admin roles to the user administrator, application administrator, and/or network administrator or similar. ► Back to the Security Trail: According EMA GMP Annex 11 – 12.3 – “Creation, change, and cancellation of access authorisations should be recorded” anyway with appropriate request forms (incl. reason for change) similar to change records (preferable as electronic forms). But the Security Trail for the user management and administration itself could be used for the recording / documented evidence that the change was executed. In addition it can be used to show during the periodic evaluation (ref. EMA GMP Annex 11 – chapter 11) that the user accounts, the group assignments, etc. do comply. Another kind of Security Trail can show the login logs and attempts, which might be useful (for open systems) for intrusion detection and security management. This can be reviewed during the periodic evaluation or if needed and critical during special security checks. We should not call this an “Audit Trail Review”, because such logs look totally different like GMP Audit Trails. For example, a hacker to an open system would not use a real user name – the IP address would be much more interesting, which can then be blocked. ✔ “Audit Trail” really needed? ► What is an “Audit Trail”? Is this a basic question? Or at least it is not a simple one. ► Let’s start with a deliberate provocation: Does a system really need to have an “Audit Trail” in general? Simple answer: NO. Although a standard sentence in any URS for a computerized system is poorly formulated that the system should have an Audit Trail functionality. Is this really correct? ► Again, basically not, if data is at minimum following a defined and compliant status control like any other (paper-based) document or record (reviewed and approved). The audit trail function itself is a luxury and comfort function. It enables the user / operator / analyst to change data in real time, ideally within a predefined time period and a predefined range. The audit trail function records automatically the old and new values (status), date and time of change, person performing the change and the reason for the change – this is indeed very comfortable. Basically this all can also be handled by a “traditional” change control record and manual print-outs and screen-shots attached to the change request – but this takes a lot of time, may cause errors and faults, and is far away from “real time” working (keyword: real-time release / operational excellence). ► But for now it is important to realize that the “Audit Trail” function is a great nice-to-have function. And the conclusion for that must be that it is very important how and in which context a GMP process owners allows the user / operator to make online / real-time changes of data. Before having computerized systems and more manual processes the process owner needed to define if changes were recorded into a machine / instrument logbook, if a change needed to be requested by the change control process, if an event entry should be recorded in the batch / laboratory records and/or if such an event would trigger a deviation record. All variants could be found nowadays. ► Beside of that we need to define first what exactly can be changed. The basic GMP documentation is defined into two types: instructions and records/reports. For sure changing pre-approved instructions must be seen as critical. We would also agree on that changing critical quality attributes (CQA) of a product would be critical. Changing critical process parameters (CPPs) might be allowed, if a Design Space and a pre-defined, approved Control Strategy would be in place. Even changing system parameters or methods might be allowed, if again such a processes is pre-defined and approved. This is at the end also a question of following a retrospective or prospective QRM approach (Quality Risk Management), without going into details here. ✔ What should be audit trailed? Or which role must be trailed? ► Coming back quickly to the “administrator” group mentioned above: Let’s assume there is a user group defined as “QC analysists” who exclusively performs QC test runs. Another group, let’s call them “application admin” is managing the analysis methods. Each method is version- and status controlled. That means that the QC analyst can only use the last approved version for a sample run, cannot modify the method before, during and after the test run, and the method must be approved by a QA role. No individual is assigned to both groups in parallel. Any change of the method must be requested by a change request. Different versions of the method can be compared. Now the magic question: Do we need (mandatory) an audit trail for the group QC analyst and secondly for the application admin group? With no doubt an audit trail or maybe it is more an automated change log would make any investigation easier, but it is not really mandatory in this case. There might be a lot of BUT and IFs, but in most of the cases it should be analysed if an audit trail (and which type) is really needed. If processes and work flows are well designed, restrictive, secured, fit for purpose it might not really required to run all data objects under audit trails. ✔ Fact #4: Data objects / sets must be version-controlled. Data objects which are configured to be tracked by an audit trail function should have such version control (V1.0 of data set) and/or status control (in review/approved). In more complex systems and data relations such version controls can cover purely the vertical or even the horizontal tracking of each data object and related data references. ✔ Audit Trail Review – do the right thing ► Let’s start with a simple statement: The term “Audit Trail Review” is not mentioned in any guideline or regulation. EMA Annex 11 states that “audit trails… should be regularly reviewed.” ✔ Fact #5: The “Audit Trail Review” contains two levels of verification: Verification of the “Audit Trail” function and verification (review) of the Audit Trail records. ✔ Fact #6: The purpose and objective of the “Audit Trail Review” (of records) is to gain knowledge (ref. ICH Q10) of the product and process and the linking (relationship) between product (CQA) and process (CPP and system parameters) and emphasizes product and process understanding. ► We might agree that this “Audit Trail Review” in the context of GMP must not be executed for the Security Trail (covered by periodic evaluation) and the Activity Trail (covered by the batch / lab. record review). ✔ The real GMP Data Audit Trail ► Now it is really time to have a look on the real GMP Data Audit Trail. Before that proper GMP Data and Records Management requires a good understanding of the overall GMP/CGMP regulations and modern Quality System, Product Development and Process Management (QbD) as defined for example in ICH Q8, Q9, Q10, and Q11. ► In very simple words there are two different Product / Process approaches, for example according EMA Annex 15: Manufacturing processes may be developed using a traditional approach or a continuous verification approach. ► We may call the traditional one the “Quality-by-Testing” (QbT) approach and the continuous verification approach “Quality-by-Design” (QbD). With the traditional QbT methodology the critical process parameters are fixed and pretty static (examples can be found in ICH Q11). On the contrary the QbD approach is based on a so called Design Space and Control Strategy, which may include a feed-back control system (or also called PAT – process analytical technology). ► The real magic between Product and Process (knowledge) comes in when the Quality Attributes and Process Parameters are mapped to each other, refer to ICH Q11 - chapter 3.1.5. Linking Material Attributes and Process Parameters to Drug Substance CQAs. Just to keep in mind, again the basic idea is have a greater output of medicines with a better quality. So products must have a defined Quality Target Product Profile and associated CQAs and CPPs, which we must understand, for new and existing products. ► Linking Critical Quality Attributes (CQA) and Critical Process Parameters (CPP) is not an easy task and it is not a one to one or linear relationship. ✔ Facing systems and analyse ► Annex 11 states that “consideration should be given, based on a risk assessment, to building into the system the creation of a record of all GMP-relevant changes and deletions…” ► If you stand in front of a system, machine, instrument etc. you may ask yourself which data is really GMP relevant and which one should be trailed (refer Annex 11: based on a risk assessment)? ► In general and as a very basic rule: Production machines (e.g. PLC) cover CPPs QC Lab instruments and systems cover CQAs On ERP (MES) level: Management of master and transaction data Electronic QMS systems: Quality System data (CAPA, training etc.) ► Relevant are foremost the CPPs, which are normally fixed nowadays in a traditional production model. Changes of the CQAs, if at all changeable, won’t be possible. On ERP or eQMS level corrections of quality, master and transaction data should be possible (mistakes will happen). ► It might be surprising if that Data Audit Trail is limited to all online and real-time changeable CPPs there won’t be a high number of these. And as such the so called Audit Trail Review will also be limited to this number of Data Audit Trails. ✔ Changes of system configuration ► First of all, yes it is correct for the CPPs, but as already mentioned these are also function of the system settings / configuration / programs or defined methods. So data impacted, created, controlled, calculated and recorded by systems must also be version and status controlled, exactly like the created data. For systems the interpretation must be that they have a proper release, change and configuration management. ► Magic question: Is it required to have a – real-time, pre-approved, lean – audit trail function for changes of the system programs by the programmer? No, because such changes must be analysed, executed, reviewed and approved by several roles and an audit trail would not be appropriate for that. ✔ Audit Trail Types ► I have defined three “Audit Trail” types:
✔ Data Audi Trail Specification ► Having a detailed view on the Data Audit Trail:
► What must happen now? There are different options or even opinions (based on criticality / severity):
► This is just an example for the Data Audit Trail process. There are also other specifications to be defined for a proper Data Audit Trail. In general a good audit trail specification covers more than 20 requirements, like for the following areas: ► Audit Trail function: General requirements for the function, e.g. date and time reference taken from network time services, logical function setup, impact of changes of master data (e.g. user name changes) etc. ► Audit Trail configuration: Requirements for the configuration settings of an audit trail like date format and time zone, configuration of which data objects should be trailed and with which type of audit trails. ► Audit Trail listing and view: Requirements for displaying, sorting, selecting audit trails with user access rights. ► Audit Trail print-outs: Controlled print-outs, Report design, etc. ► Audit Trail Analysis: Automated analysis for “Audit Trail Review” (exception report) ✔ Specification of Audit Trail Review ► When we have specified the Audit Trail above there should also be a specification for the “Audit Trail Review”. It might be that some persons define the review as a manual and paper-based process. It should not be like that. The Audit Trail Review should be automated (refer PIC/S PI 041 – exception report). That means that the audit trail review, if defined to be needed, should be done by the system or even systems. *This article was originally posted on www.linkedin.com
 "To best support your data integrity needs for your regulated infrastructure, software and systems within IT, Automated MES and interfaced SAP or Oracle ERP, SaaS Public Cloud Projects and BI / BW, the FDA external audits want to examine the following as part of each respective audit for your Prospective, Concurrent or Retrospective External CGMP Process Validations": ✔ A comprehensive FDA audit investigation should include: ► Detailed investigation protocol and methodology; summary of all laboratories, manufacturing operations, and systems to be covered; justification for anything to be excluded. ► Interviews of current and former employees to identify the nature, scope, and root cause of data inaccuracies. Should be conducted by a third party. ► Assessment of the extent of data integrity deficiencies. Identify omissions, alterations, deletions, record destruction, non-contemporaneous record completion. Describe all operations with data integrity lapses. ► Comprehensive retrospective evaluation of the nature of the data integrity deficiencies. Qualified third party with expertise specific to firm’s breaches should evaluate the lapses. ✔ The FDA will evaluate your audit compliance with Current Good Manufacturing Process (CGMP) based on both inspection reports and evidence gathered by FDA investigators for the following criteria measured against your internally defined procedures for the following: qualification, validation, data governance, data integrity and data management: ► An established development or process methodology, such as SDLC, Agile or a combination of the two with a transparent resource driven project plan for all the relevant validation project phases for respective CGMP Process Validations. ► High level risk assessment. ► A written and approved Infrastructure Qualification Plan, supporting detailed Risk Assessment to deal with Computing Facility Hazards by way of ensuring the required risk controls are in place; supported by an Infrastructure Qualification Report. ► A written and approved Validation Plan (and Test Plan if included which can also clarify the classification of deviations and phased acceptance criteria). ► Documented user requirements for both functional and non functional aspects. The solution blue print should also be present with the corresponding business process maps with clearly defined operational role involvement within the associated process swim lanes. ►Documented integrated functional design specifications with supporting integration tests and system tests. ►Documented architecture design specifications clarifying any infrastructure for both normal and virtualized set ups. ►Documented testing protocols for OQs and UAT project phases (PQs), and evidence of reviews with objective test results included. ►Documented evidence of installation protocols (installation guide and the configuration guide) and evidence of test reviews and approvals. The inclusion of Test Summary Reports from either your inhouse IT Team or your preferred Partners and Vendors are most important when establishing each new IQ base line. ► Clarification on the segregation of duties. ► Migration approach and procedures and verification scripts. ►A written and approved Validation Report; this can also be supported by an OQ Report and UAT Report and a test closure report. ►Complete and documented traceability (from User requirements to testing which make reference to the functional specification, identified risks, OQ and UAT test scripts via the related ID numbers). ► Documentation of responsible approvals - Normally highlighed in the Validation Plan. ► A defined maintenance and change control process, including an infrastructure and data risk analysis. ► Documentation of changes with associated IQs, impact of those changes, risk assessment of those changes, and periodic monitoring. ► An effective vendor management process. ► System security, roles and privileges, network access, firewall access, Public cloud access for the various classified data types if the information is also held on a public cloud of a given SaaS solution - for more infomation please see my article on the "Public Cloud." ► Supplier Assessments. ► When dealing with CAPAs due to FDA observations, 483s and FDA warning letters, complaints, deviations, out of specification items, and non compliance on your business processes, it will help you to plan for "Sustainable Change" regarding your proposed innovation improvements which are under both scrutiny and inspection.  ► Clarifying the all important data integrity management for the introduction solutions mentioned at the beginning of this article via the evolutionary Electronic Records and Electronic Signatures CFR Part 11 regulatory demands: An official definition of “data integrity” is not found in the regulations. FDA and other regulatory authorities expect that data will have attributes described in the acronym ALCOA. As applied to GMP, that means data is expected to be: ✔ Accurate: Data must be accurate. Where appropriate, correctness should be second-person verified. Data accuracy extends, for example, to data/information that is presented in multiple locations — such as an equipment log, laboratory notebook, and electronic chromatography records — where data should be in agreement. ✔ Legible: Data and results must be legible/readable. Electronic data must also have the capability to be made human-readable. ✔ Contemporaneous: Thus, data is recorded at the time of the event/action, not transcribed at a later date. Data is not transcribed from sticky notes or scrap paper to the official documents, such as batch records or laboratory notebooks. ✔ Original: Original data is similar to “raw data.” FDA’s opinion: “Original record: Data as the file or format in which it was originally generated, preserving the integrity (accuracy, completeness, content and meaning) of the record, e.g. original paper record of manual observation, or electronic raw data file from a computerized system.” ✔ Attributable: This term requires the ability to determine who logged into the system, who collected the data, when it was collected, from which instrument it was collected, who processed the data, and who made any data modification or data manipulations. Use of shared passwords renders makes it impossible for the reviewer to attribute the data to a specific person. ► Further clarification on the data integrity concepts are as follows: ✔ Metadata: ► Contextual information required to understand data. ► Structured information that describes, explains, or otherwise makes it easier to retrieve, use or manage data. For example: date/time stamp, user ID, instrument ID, audit trails, etc. ► Relationships between data and their metadata should be preserved in a secure and traceable manner. ✔ Static vs. dynamic records ► Static: fixed data document such as a paper record or an electronic image. ► Dynamic: record format allows interaction between the user and the record content such as a chromatogram where the integration parameters can be modified. ✔ Backup data and archiving and data retention ► True copy of the original data that is maintained securely throughout the records retention period. ► Should include associated metadata. ► Not backup copies that may be created during normal computer use and temporarily maintained for disaster recovery to ensure business continuity. ✔ System validation Does each workflow on our computer system need to be validated?: ► Yes, a workflow, such as creation of an electronic MPCR, is an intended use of a computer system to be checked through validation. ► If you validate the computer system, but you do not validate it for its intended use, you cannot know if your workflow runs correctly. ✔ Audit Trail ► Secure, computer-generated, time-stamped electronic record that allows for reconstruction of events relating to the creation, modification, or deletion of an electronic record. ► Chronology: who, what, when, and sometimes why of a record. ► CGMP-compliant record-keeping practices prevent data from being lost or obscured Audit trails capture: ► Overwriting ► Aborting runs ► Testing into compliance ► Deleting ► Backdating ► Altering data How often should audit trails be reviewed?: ► For audit trails that capture changes to critical data, FDA recommends review of each record before final approval of the record. Audit trails subject to regular review should include changes to: ► History of finished product test results ► Sample run sequences ► Sample identification ► Critical process parameters The FDA recommends routine scheduled audit trail review based on the complexity of the system and its intended use. Final point on the CFR Part 11: “Audit trails” are frequently cited in FDA enforcement actions. It is important to remember that audit trails in electronic records are the equivalent of the “line-out, initial and date, explain” process used to identify and correct mistakes made in paper records. In the absence of appropriately configured and enabled audit trails it is impossible for a reviewer or auditor to ensure the data is valid and has not been altered or deleted. Warning letters have been issued for permitting conditions to exist where data may be changed or deleted. ► The supporting IT Infrastructure Qualification Report (QR) which comes before the VR will close out the qualification activities for the given Infrastructure. This will stipulate the formal handover to operational use even if the IT infrastructure may be in operation prior to the formal handover. All further maintenance (changes) must be maintained in the change control system. ► In summary, the Infrastructure QR includes: ✔Qualification deliverables, standard operating procedures and document references ✔Deviation and unexpected results during the qualification ✔Any changes that have been made with CR references ✔Outstanding issues and corrective actions ✔Qualification status ► The Validation Report (VR) will summarize the results of the Validation activities and give a status for each acceptance criteria described in the Validation Plan. The VR concludes on the validation status of the identified scope. The VR further acts as the release form for the GxP identified infrastructure and combined application usage within the production environment, and helps to make future validations less complex. The VR presents the final validation status of the system, and requires official signature and approval. ► Any open issues and deviations will be documented and where applicable mitigation or remediation measures will be defined with responsible(s) and due dates. The Validation Report should only be approved if no high critical deviations are outstanding. ► And the VR includes: ✔Validation deliverables, standard operating procedures and document references ✔Deviation and unexpected results during the validation ✔Any changes that have been made with CR references ✔Outstanding issues and corrective actions ✔Validation status GMP Audits also refer to the following documents: ► Relevant management system documents and records ► Applicable standards ► Site master file ► Registration files ► Guidelines ► SOP’s and WKI's ► Protocol or Project specific requirements ► Records of the company in relation to complaints and recalls ► Customer satisfaction ratings ► Supplier performance metrics ► Corrective and preventive actions (CAPAs) ► Nonconformance reports (NCRs) ► Management reviews ► Previous Internal and external audit findings can be used ► These guidelines are fully supported and evaluated by "GAMP 5". A GAMP Assessment may allow you to refer to previous GAMP versions depending on the level of both your Infrastructure and Application maturity. The FDA will use the following enforcement tools when required: ✔ Regulatory Meetings ✔ Injunctions ✔ Consent Decrees ✔ Import Alerts ✔ Seizures ✔ Warning Letters ✔ Untitled Letters ✔ and more... Should personnel be trained in detecting data integrity issues as part of a routine CGMP training program? ► Yes, detecting data integrity issues is consistent with the CGMP requirements for personnel qualifications. ► Personnel must have the education, training, and experience, or any combination thereof, to perform their assigned duties. Is the FDA investigator allowed to look at my electronic records? ► Yes. All records required under CGMP are subject to FDA inspection. See the following FDA guidance, circumstances that constitute delaying, denying, limiting or refusing an inspection: http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM360484.pdf Finally, once the FDA audit findings are communicated, you must put in place a proactive management strategy that includes the details of your global corrective action and preventive action plan. Your strategy should include: ► A detailed corrective action plan that describes how you intend to ensure there liability and completeness of all of the data you generate, including analytical data, manufacturing records, and all data submitted to FDA. ► A comprehensive description of the root causes of your data integrity lapses, including evidence that the scope and depth of the current action plan is commensurate with the findings of the investigation and risk assessment. Indicate whether individuals responsible for data integrity lapses remain able to influence CGMP-related application data at your firm. ► Interim measures describing the actions you have taken or will take to protect patients and to ensure the quality of your drugs, such as notifying your customers, recalling product, conducting additional testing, adding lots to your stability programs to assure stability, drug application actions, and enhanced complaint monitoring. ► Long-term measures describing any remediation efforts and enhancements to procedures, processes, methods, controls, systems, management oversight, and human resources (e.g., training, staffing improvements) designed to ensure the integrity of your company’s data. ► A status report for any of the above activities already underway or completed. Bonus materials: ► To understand "Agile," please check out my published article by clicking the link. ► To understand how best to deal with "Embedded Software." ► To be well informed about "SAP BI." ► Remember to produce, maintain, and train your staff on all relevant SOPS and WKIs. ► Insights into the world of Infrastructure Qualifications which clarifies the differences between Qualification, Validation and Control. ► Please see my popular article on " UAT Entrance Acceptance Criteria." *This article was originally posted on www.linkedin.com
 1. PILOT Scale Development Phase: Literature Search, Active Sourcing and Evaluation, Active and Innovator Product Purchasing & Testing, Formulation and Analytical Method Development, Process Optimization and Scale Up, Analytical Method Validation & Transfer, Pilot Stability & BE Study, Green flagging from Internal Regulatory and Patent Cell. 2. PIVOTAL Scale Execution Phase: Process Qualification-Pre Exhibit Batch, Pivotal Production, Pivotal Stability and BE Study, Pre Submission Auditing within F&D, ARL, Production, QA, QC, RA, ANDA Submission to FDA, Validation of 3 Consecutive Batches, Review of ANDA by FDA, Approval of ANDA, Mfg Facility inspection by FDA, Re-inspection of Mfg Stds biannually by FDA. This "S" Model of ANDA covers major elements of Project Management: 1. Integration & Scope Management 2. Human Resource & Procurement Management 3. Time & Cost Management 4. Quality Risk Management 5. Communication Management *This article was originally posted on www.linkedin.com
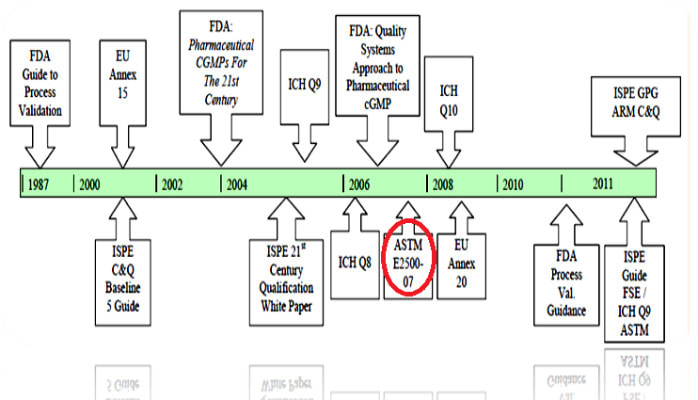 Genesis of Verification Industry… 2001 - ISPE Baseline Guide 5 establishes impact assessment model 2004 - FDA approaches ISPE with concerns on current practices
2007 – ASTM E2500 published Why ASTM standard??? FDA Process Validation Guidance 2011 refers to implement this standard for facility, system and equipment qualification purposes. ASTM E 2500, What is it??? “A risk-based and science-based approach to the specification, design, and verification of manufacturing systems and equipment that have the potential to affect product quality and patient safety.”
Pharmaceutical and bio-pharmaceutical manufacturing Systems:
Specification Design Verification Lifecycle Alternative to ISPE Baseline Guide Vol 5 Commissioning & Qualification
Risk Assessments by Subject Matter Experts (SMEs)
“It is estimated that validation can add up to 25% of the total installation cost for new facilities.” This standard guide avoids traditional terminology to reduce verification costs and project time using new risk based approach. E 2500 – puts focus on Critical areas that affect
 Traditional Terminology 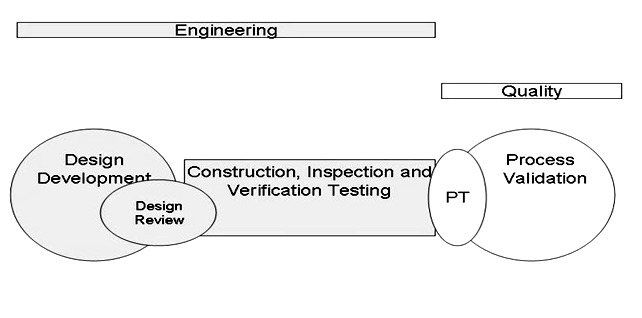 New Terminology Qualification -Traditional vs. Risk Based Approach
So, ASTM E 2500 Is… A standard approach for validating equipment, facilities, processes
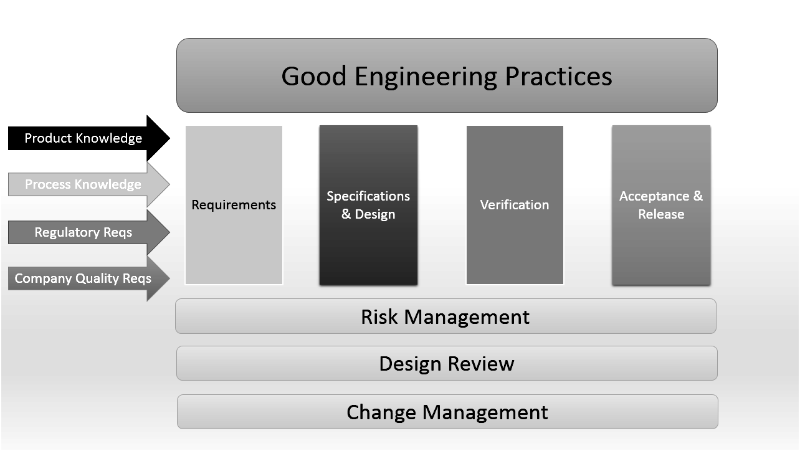 ASTM E2500-07 lifecycle phases ASTM E2500 Key Concepts…
Benefits of Implementing E 2500… LEAN manufacturing systems - Remove waste Elevate our industry to more knowledge, better understanding of our manufacturing systems - Data, PAT, Design Space Focus on what’s important (Critical) - More is not better Better technical understanding - (Subject Matter Experts) Less waste & repetition - Use vendor doc’s How best you can apply ASTM E2500??? Step 1: Develop Supporting Processes
Step 10: GMP Operation and Change Management References *This article was originally posted on www.linkedin.com
|

© 2016-2023 GMP GEEK : a first pharmaceuticals gmp practitioner weblog from Bangladesh
